PublicSequenceResource
Coordinator: Thomas Liener
One recurring idea is to create an uploader where raw data from a sequencer (long reads and short reads) is loaded onto a backend and mapped using traditional tools as well as the variation graph/pangenome tools. Next a visualization is generated of the viral strain in comparison with data we already have in the database. Furthermore, phenotypes that we have and metadata can be presented at the same time, to show how this viral strain relates to other strains, geo info, clinical info, treatment info - anything that we have and that can be linked out. Obviously the uploaded data becomes part of the whole.
The justification of such an uploader is easy. Currently there is no system that handles ontologies well. Currently there is no system that allows for on-the-fly analysis of raw data.
Mind, this is a pretty large project! But if we split it into small parts where each group owns subsections we should be able to put it together and make a working prototype. When the full application works we can improve after the BioHackathon and encourage data providers to add their material. As a BioHackathon we can get a high impact paper out of such a project though that is not the primary goal.
We can discuss subtasks here and ask for group coordinators for each subtask to work out what needs to be done? Subtasks we identify:
- Uploader with authentication, uploading fastq or BAM, add known (clinical) phenotypes. Study usage of Phenopackets as standard for phenotypic data submission. Going the other direction, this may be useful: omopomics
- Create workflow for traditional analysis (coordinator Michael R. Crusoe)
- Create workflow for vgtools (coordinator Michael R. Crusoe)
- Run workflows in cloud/HPC (coordinator Michael R. Crusoe)
- Store results in persistent storage (coordinator Michael R. Crusoe)
- Metadata and ontologies (interim? coordinator Thomas Liener)
- Define and query linked data (wikidata) (interim? coordinator Thomas Liener)
- Create visualization (coordinators Josiah Seaman and Simon Heumos)
- Create output website
- Coordinate with existing efforts (e.g. NextStrain, ELIXIR, others) to be able to port data back and forth! Ben Busby -- if anyone has contacts that they want to share -- please do so in slack and tag me!
All items (1-9) Vanessasaurus
Does that sound reasonable? Other tasks may be
- Deploy graph store, database, IPFS (coordinator Pjotr Prins)
- Deploy cloud/HPC workflow runner (coordinator Pjotr Prins)
- Deploy web interfaces (coordinator Pjotr Prins)
The Galaxy team already has put some things in place and we may be able to collaborate on this. Galaxy team, wdyt?
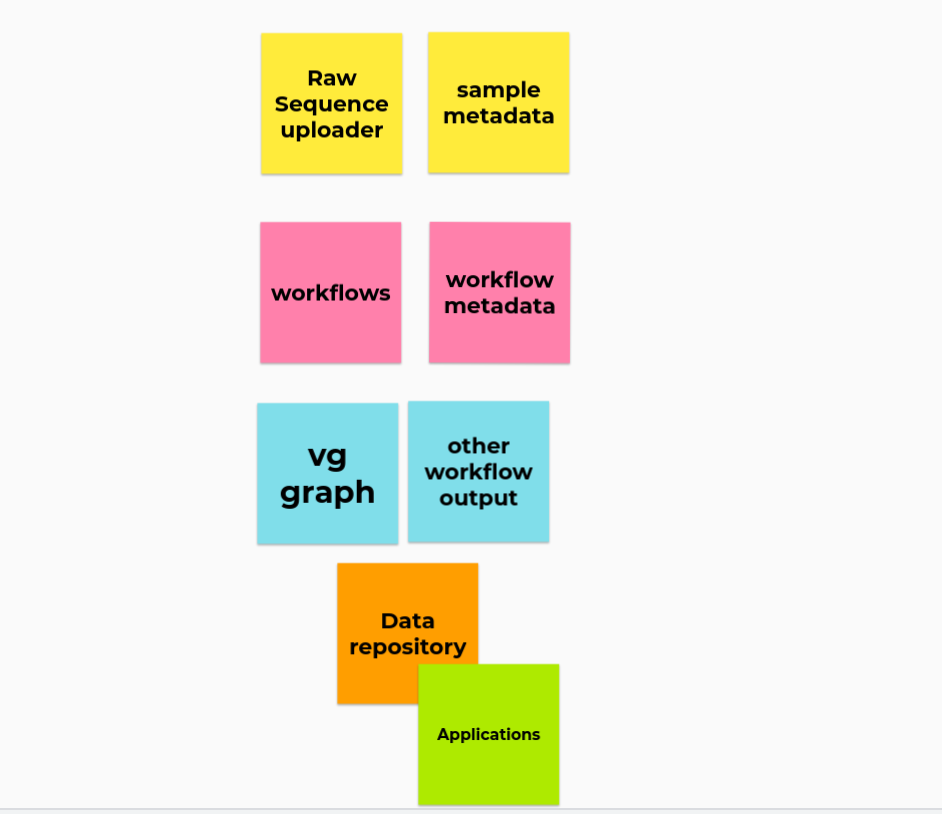
This should define the pieces and what BH groups might takes on which topic. The change compared to the plan above is that we aim for a data repository as goal, while leaving the output website to "consuming applications" if they wish. (A consuming application could also be a public resource I guess, does that make sense?).
- (Yellow) Raw sequence up-loader (Pjotr, Workflows) and sample metadata (Ontology, FAIR Data)
- (Pink) Workflows and workflow meta data (Workflows, Workflow hub)
- (Blue) Workflow output (Workflows, Pangenome, ...)
- (Orange) Data repository: Searchable data access (Ontology, FAIR Data)
- (Green) Applications: Different applications and BH subgroups could consume the data (All interested Applications)
Raw sequence up loader (coordinator Peter)
- Provide a way to easily and securely upload data
- As POC a command line tool for submission (e.g. python) would be sufficient https://github.com/arvados/bh20-seq-resource/
(Sample) meta data (coordinator Thomas)
- Define a meta data model to go along with a raw sequence submission.
- Provide a way to validate a metadata file against the defined model (e.g. python) so it could be included/combined with the raw sequence up-loader http://covid-19.genenetwork.org/
Workflows (coordinator Tazro with support Erik+Michael)
- Take raw data submission and its meta data and execute workflows
Workflow Data HUB
- What do we need to make the data repository searchable etc?
https://github.com/hpobio-lab/viral-analysis/tree/master/cwl/pangenome-generate
- Fasta uploader + (some minimal) metadata, fed into workflow to create a VG