Move filter after subsampling #814
Conversation
|
I tested this workflow while running some builds for African countries by starting with the full unaligned Nextstrain GISAID sequences on S3 with a three-part subsampling scheme that includes focal sequences for a country, regional context from Africa, and global context from outside of Africa. By setting the inputs entry point to
The sanitize metadata script is obviously quite slow here, but this is a one time cost that subsequent builds for other countries don't incur. The focal and regional subsampling take ~10 minutes and the global context takes ~25 minutes. These commands only output strain lists and sequences get extracted by a later augur filter command that runs in ~15 minutes. Most importantly, the subsampling logic doesn't use priorities, so we can completely skip alignment of the full GISAID database. This workflow ran in ~3 hours instead of the ~15 hours it would have taken if we included alignment. |

|
@huddlej great to see this run time visual for an end-to-end build! Curious, what did you use to generate this? |
|
I used @tsibley's Snakemake run stats viewer. |
|
@jameshadfield mentioned the idea that we could build on these changes such that we don't need to fake that the inputs are aligned, but instead allow users to define a |
|
Here are stats for a subsequent Egypt build after sanitizing metadata has completed (Egypt has a similar number of genomes as Ghana):
This build took just under 2 hours. |

There was a problem hiding this comment.
This is great @huddlej, thanks for tackling this. I've not run this, but have compared the DAGs and reviewed the code. (I realise this is still a draft PR so you've not covered everything).
I wrote responses to your in-line comments but in this PR they're showing up as individual comments for some reason. I think there are a few additions that are needed or at the least will help reduce papercuts:
Can no longer start from filtered inputs
The official nextstrain profile, some tests and the "example_global_context" build start from filtered inputs. Additionally the docs discuss filtered inputs in multiple places.
P.S. I'm going to make a PR on top of this one to remove the preprocess profile, so you can ignore those if you wish. Done, see #823.
Config warnings
The schema should remove filtered as a valid input which will prevent those configs from running. Additionally, if a user defined a config setting filter → {origin} then we should exit and notify them that this is no longer possible.
|
Thank you for looking through this, @jameshadfield! It was in the back of my mind to go back through all the configs, example, builds, and docs to remove references to "filtered", but I should have written it out. We just found out today that Daniel Bridges (from the Zambia PATH group) would like to use the input-specific filtering option of the ncov workflow that we talked about dropping yesterday. The ability to apply different filters per input was an important feature. There isn't a way in the current PR to support per-origin filtering by number of valid nucleotides (min_length) or a way to skip the diagnostic for specific inputs. The presence of the origin columns in the metadata would help with the second case. If we annotated number of valid nucleotides in the metadata instead of the sequence index, users could use the |
| """ | ||
| augur filter \ | ||
| --sequences {input.sequences} \ | ||
| --metadata {input.metadata} \ |
There was a problem hiding this comment.
We could support input-specific filters as long as the metadata include columns like length or ACTG or valid_bases and then users could define a query that filters based on metadata input column like:
--query "(gisaid == 'yes' & length > 5000)"
If we update the alignment step above to use Nextclade instead of Nextalign, we could get the annotation of the number of gaps, etc. in a TSV to merge with metadata to allow the query above. Alternately, it would be simpler to start with augur index output and another rule+script that merges the metadata with the index.
@jameshadfield points out that we could generate this query based on the existing filter config if users provide an input-specific filter and add that query to the existing augur filter. To do this we would loop through all "origins" defined in inputs and construct a query string with a component per origin that pulls from the config. For a config like the following:
inputs:
- name: gisaid
# ...snip...
- name: aus
# ...snip...
filter:
min_length: 10000
aus:
min_length: 5000the resulting query would be something like:
--query "(gisaid == 'yes' & length > 10000) | (aus == 'yes' & length > 5000)"
Because we generate this query from Snakemake, we can call the length field whatever we like and change its source in the future without breaking the workflow. We should consider calling this field a private name like "_length" or something that won't collide with existing metadata columns.
There was a problem hiding this comment.
Thinking about this more now, this solution only addresses the specific use case of filtering by length. We've technically allowed users to define any valid filter key for any origin and we've documented four specific options in the configuration guide and two of these in the multiple inputs example.
Instead of figuring out how to support each of these individually or collectively, we could revisit the use case. Here's an attempt at the user story we're trying to support:
James has 20 new likely Omicron genomes from his city that just came off the sequencer and he wants to know how they place in the phylogenetic context of other sequences from the city and the country. He configures a Nextstrain build with inputs including the full GISAID database and his local sequences and metadata. He wants a high-quality set of GISAID data for his phylogenetic context, but he wants to keep all local sequences in his analysis regardless of their quality. He sets stringent quality filters for GISAID data, applying a minimum genome length of 27,000 valid nucleotides and filtering out any genomes that do not pass all of Nextclade's quality control annotations.
From this user's perspective, we've got the current implementation a bit backwards; James wants to keep all of his inputs by default (with reasonable subsampling) and only wants to apply filters to specific datasets. We currently require the opposite approach where we apply more stringent filters by default and require users to manually relax those filters.
There was a problem hiding this comment.
I do think having a more stringent default is likely good - for users less clear on what's going on, I think reducing the odds they take away big messages from poor quality genomes is fair. However, it should also be as straightforward as possible to relax these if you actively are choosing to include worse-quality genomes (which is a very fair thing to do - just really important you know you've done it!).
I think if most users are interested in QC metric filtering, we could try to get this from Nextclade early on (as you suggest) & support quite a few different filtering options - but not "anything". I guess we also ideally would want for user-datasets to go through this, but not to try and generate this for GISAID - preferring to wait until after subsampling (though those within Nextstrain can use the files we've generated already with this info).
I like the idea of putting this query together from the config as suggested above.
|
@jameshadfield Based on our conversations (above and offline), I've made the following changes to this PR:
The most recent commits add two rules to the workflow including creation of a smaller sequence index after subsampling and merging of that index with the metadata to get "private" metadata fields with sequence content information. We could also decide to open this up a bit and make this annotated metadata more standard throughout the workflow, so it could be queried against more generically. For a builds config like the following: The filter step dynamically builds a query like this: The diagnostics script also runs with an additional argument of For all other input-specific filters, the user gets an error message saying that these are not supported. We'll need to test this workflow a bit more, after such major surgery. I will run some full Nextstrain builds on rhino and also some builds that use priorities, to get a better sense of where other bugs might exist. If you have other tests you'd like to run (e.g., with your own local data, etc.) that would be helpful, too. |
|
I ran two full Nextstrain builds in parallel (Africa and Europe) on the cluster with this PR. The builds ran in ~3 hours. One of the longest steps was the sanitize metadata step, as shown below.
In a separate PR, we should consider adding a boolean flag for each input to allow skipping the sanitize steps. Like: inputs:
- name: gisaid
metadata: "s3://nextstrain-ncov-private/metadata.tsv.gz"
aligned: "s3://nextstrain-ncov-private/sequences.fasta.xz"
sanitize_metadata: false
sanitize_sequences: falseEveryone else who uses GISAID-sourced data will still need these early steps, but the Nextstrain builds would run faster without them. Next, I will run some similarly-sized builds with priorities enabled for subsampling. |

Starts to reorganize the workflow such that we only need the sequence index when subsampling with priorities and we only filter after the subsampling, alignment, and mask steps. Related to #810
These should be small enough, it isn't worth the extra effort/complexity to compress.
Reverts inputs for pangolin annotations and augur ancestral to match the current workflow, so these tools gets unmasked sequences. This change allows pangolin to do what it needs to internally without preemptively throwing out information and keeps mutations from masked sites as "decorations" on the tree even though these sites are otherwise ignored during the analysis.
Maintains support for two input-specific filter settings: `min_length` and `skip_diagnostics`. To support the minimum length filter, this commit adds a sequence index rule to the workflow before filtering, a rule and script that annotates the metadata with sequence index data, and custom query parameter logic in Snakemake to build a filter query for each input based on the minimum length given. Additionally, this commit allows users to skip diagnostics for specific inputs. When there is only one input, the top-level `skip_diagnostics` parameter takes effect. When there are multiple inputs, the workflow will look for input-specific requests to skip diagnostics. The diagnostics script now optionally takes a list of input names whose records should be skipped and filters these prior to other diagnostic logic.
Alerts the user when input-specific filters are no longer supported, so their workflows don't silently produce unexpected results.
Handle cases where `skip_diagnostics` is not defined for a build and where only one input exists for we can't filter metadata by the input name.
|
@jameshadfield The open builds we queued up yesterday ran without errors in just over 2 hours! 🎉 Edit: For comparison, the open build I just ran for the "allow numbers in build names" PR took a little over 4 hours. |
There was a problem hiding this comment.
Looks great - I've looked through the code again and the successful open builds indicate that this works in practice.
The current actions look like this:
- Ncov-ingest (GISAID/GenBank) -> trigger ncov preprocessing pipelines -> trigger ncov rebuild pipelines
And after this PR they look like:
- Ncov-ingest (GISAID/GenBank) -> trigger ncov rebuild pipelines
Note that the companion PR ncov-ingest #255 is needed here which stops ncov-ingest trying to trigger the (non-existent) preprocessing pipeline.
@huddlej and I plan to merge this tomorrow (Wednesday Seattle time). The schedule for ncov-ingest runs is:
- 14h07 UTC daily for GISAID which is 10h07 Seattle
- 12h07 UTC daily for GenBank which is 08h07 Seattle
I propose we use the following steps to get this out:
- Cancel the schedule ncov-ingest action, assuming they have started running (see above for start times)
- Merge this PR
- Merge the companion PR ncov-ingest #255
- Manually trigger the ncov-ingest actions “GISAID fetch and injest” and “GenBank fetch and ingest” (if before 10h07 Seattle, then we could let the cron-scheduled GISAID action run instead).
- Monitor the ncov-ingest actions (no changes here except the repository dispatch trigger). These take around 3-5 hours.
- Check that the ncov rebuild pipelines are correctly triggered from step 5.
- Check the ncov pipeline works as expected (c. 2 hours runtime) and updates the correct files on S3:
- (GenBank) Builds updated at
s3://nextstrain-data/files/ncov/open/{global,africa,...}/* - (GenBank) Datasets updated at
s3://nextstrain-data/ncov_open_{global,africa,...}.json+ sidecar files - (GISAID) Builds updated at
s3://nextstrain-private/{global,africa,...}/* - (GISAID) Datasets updated at
s3://nextstrain-data/ncov_gisaid_{global,africa,...}.json+ sidecar files
- (GenBank) Builds updated at
- Remove the no-longer-used filtered sequences file from S3
- (GenBank)
s3://nextstrain-data/files/ncov/open/filtered.fasta.xz - (GISAID)
s3://nextstrain-private/filtered.fasta.xz
- (GenBank)
| @@ -71,7 +70,6 @@ This means that the full GenBank metadata and sequences are typically updated a | |||
| | Full GenBank data | metadata | https://data.nextstrain.org/files/ncov/open/metadata.tsv.gz | | |||
| | | sequences | https://data.nextstrain.org/files/ncov/open/sequences.fasta.xz | | |||
| | | aligned | https://data.nextstrain.org/files/ncov/open/aligned.fasta.xz | | |||
| | | filtered | https://data.nextstrain.org/files/ncov/open/filtered.fasta.xz | | |||
There was a problem hiding this comment.
Reminder to us to remove this file after the first successful run (and the corresponding GISAID file as well)
|
Thank you for the changes! They all make a lot of sense and we're seeing a much faster run on our end as well. Our set up is similar in that we download and pre-align all data from GISAID at night, and use the aligned fasta (which used to be the filtered fasta before changes in this PR) plus user provided data as the starting point for all our tree builds. All our tree build has priority based subsampling (builds.yaml and no changes to the main workflow), and the longest step now is the index after combining all inputs (with 6+ millions GISAID sequences. great visualization tool btw). |

|
Hi @danrlu! The index step only happens when you need priority-based subsampling. You don't see the index step in any of the examples above because I avoided subsampling with priorities. The priority calculations use the number of Ns per sequence from the sequence index to penalize/deprioritize low quality sequences. As long as your Now that you mention it, I'm realizing that we don't need the full sequence index for anything else in the workflow. We currently require it as an input for the "extract subsampled sequences" rule (to get focal sequences to compare to the whole database), but it doesn't get used. seqtk would be a much better choice for the current workflow's need. Would you want to submit a PR to replace |
|
Great tips John, thank you!! Let me swap in |
|
The testing run 2 posts above was with (Column headers are based on lh3/seqtk#47) The good news is with 6.4 million sequences, the index step went from 1h50min down to 27min, The bad news is With One work around is to count only capital letters with Comparing Side notes: I also tried The counts for The only other tool I know that may also do counting is https://bioinf.shenwei.me/seqkit/usage/ but I'm not too familiar with it. |





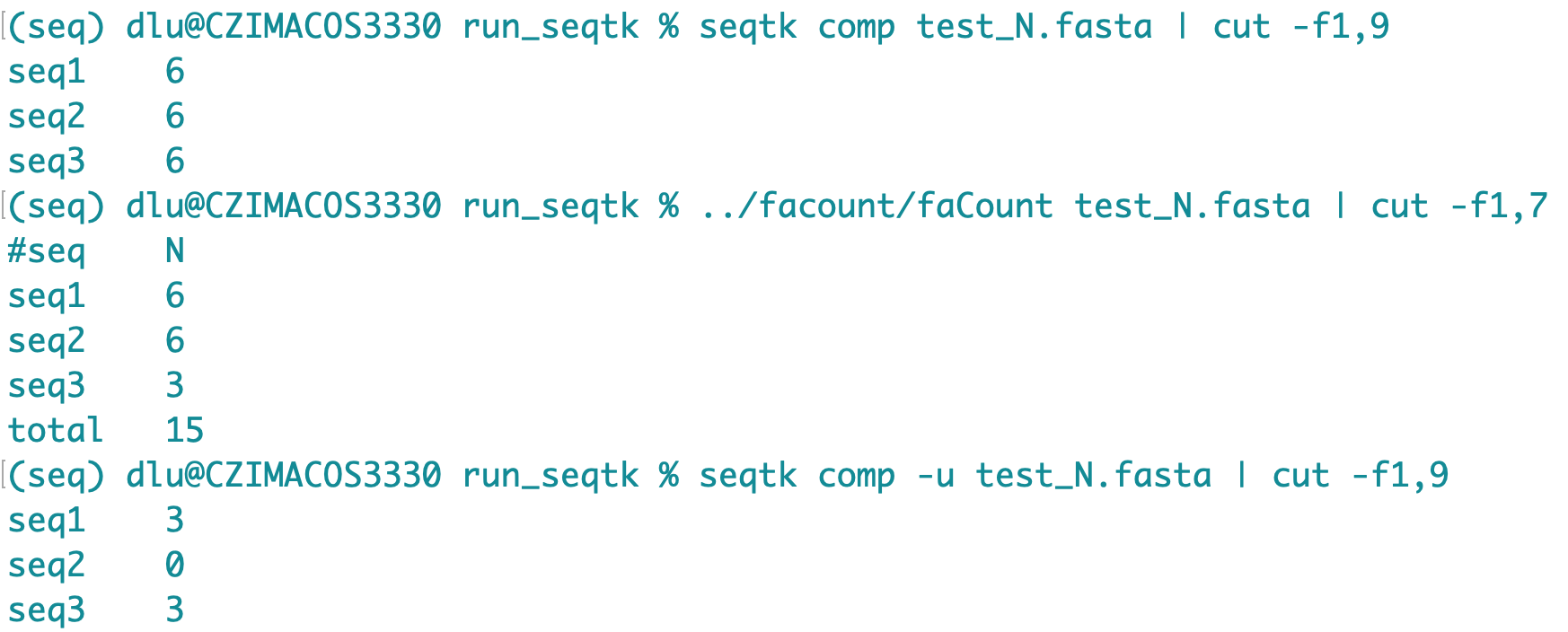
|
@danrlu Wow, thank you for the detailed analysis! Isn't this always the way with bioinformatics (or maybe just computers) that a conceptually simple task gets complicated quickly? I had a quick look at Since we already sanitize all sequences passed to the workflow, we could force all sequences to uppercase during that step and use the Some other long-term solutions could include:
The Nextclade approach might actually be ideal in that its already a parallelized C++ implementation and we already have an approach in ncov-ingest to cache alignments (and the corresponding metadata like number of missing bases). We could easily modify the priority script to look for Ns in other columns like the I'll continue this conversation with the rest of the team, but I'm curious about your thoughts on these proposals, too, and how they align with what you team has been thinking internally. |
|
Hahaha so true! It's never as straightforward as planned. With We do tree build in 2 steps: Step 2: we run Nextstrain tree builds combining user input data and the pre-processed public data from the first job. This can be done many times for many users. So anything we can stash into the first job will save us time across all tree runs. Per the potential solutions for index, I think for small tree jobs, having less package/repo dependencies is nicer and cleaner, but for large dataset, the speed matters more. Before this PR merge, step 1 takes 11hr, step 2 takes 7-8hr. So we pretty much wait a day until new data gets on to the tree. I was following this PR nextstrain/ncov-ingest#237 and thinking to do something similar, but we currently don't have any Nextclade components in our workflow and I have not looked into how Nextclade works. Per what you were describing, it sounds like we could just use Nextclade for our first job instead of |
Use aligned sequences as the aligned sequences input, rather than pass off unaligned sequences as the aligned sequences input. This should be inconsequential to workflow behaviour or results, but it makes the config a bit more straightforward and less confusing. In a quick dig thru history, it seems like ncov-ingest's aligned.fasta.xz was not _quite_ available when we first switched our profiles to use an "aligned" input instead of a "sequences" input. The original use of "aligned" with unaligned sequences was driven by run time concerns and related to the move of the filtering step after the subsampling step and move of the "preprocess" steps from this workflow (ncov) to ncov-ingest.¹ ¹ <#814> <#823>
Use aligned sequences as the aligned sequences input, rather than pass off unaligned sequences as the aligned sequences input. This should be inconsequential to workflow behaviour or results, but it makes the config a bit more straightforward and less confusing. In a quick dig thru history, it seems like ncov-ingest's aligned.fasta.xz was not _quite_ available when we first switched our profiles to use an "aligned" input instead of a "sequences" input. The original use of "aligned" with unaligned sequences was driven by run time concerns and related to the move of the filtering step after the subsampling step and move of the "preprocess" steps from this workflow (ncov) to ncov-ingest.¹ Resolves <#1054>. ¹ <#814> <#823>
This commit removes the rules `mutation_summary` and
`build_mutation_summary`, both of which called the removed script
`scripts/mutation_summary.py`.
I was looking into updating the script to work with Nextclade v3 outputs
but it seems like the rules using the script are not actively used in
the Snakemake workflow!
In Feb 2021, the script and rule `mutation_summary` were added and the
only use of the output `results/mutation_summary_{origin}.tsv.xz`
was to be uploaded it as an intermediate file of the builds.¹
In Jan 2022, the upload of the intermediate file was removed² and I
cannot find any other uses of the file so the rule `mutation_summary`
seems unused.
In March 2021, the rule `build_mutation_summary` was added to support
the rule `add_mutation_counts`.³ However, `add_mutation_counts` was
later updated to use `augur distance`⁴ which does not require the
mutation summary output. I cannot find any other uses of the output file
`results/{build_name}/mutation_summary.tsv`, so the rule
`build_mutation_summary` seems unused.
¹ <#562>
² <#814>
³ <3b23e8f>
⁴ <d24d531>
Description of proposed changes
Reorganizes the workflow to start subsampling as soon as possible without inspecting sequences. Specifically, this PR:
--exclude-allflag for augur filter)min_lengthandskip_diagnosticsby building a "min length" filter query for each input (applied as a--query) and adding a--skip-inputsargument to the diagnostics script to allow skipping inputs based on named metadata columns for each inputalignedinputs for sequences even though they are not aligned (to skip right to subsampling)Breaking changes include:
min_lengthandskip_diagnostics. The workflow prints an error to users when they try to run the filter step with any other input-specific parameters.The upshot of these changes is that the most common use case (subsampling without priorities) can run much faster from prealigned sequences by avoiding construction of a sequence index, filtering the full sequence database, and making multiple passes through the full sequence database during subsampling. When subsampling with priorities, the workflow will build a sequence index, but it will only extract subsampled sequences for the focal data. This should still be much faster than the current implementation.
Additionally, users can skip alignment completely, if they don't use priority-based subsampling, by passing their sequences to the workflow as
alignedinputs.Here is an example workflow DAG from this PR with no priority subsampling:
Here is the corresponding DAG with priority subsampling:
Related issue(s)
Fixes #810
Fixes #807
Testing
Release checklist
If this pull request introduces backward incompatible changes, complete the following steps for a new release of the workflow:
docs/src/reference/change_log.mdin this pull request to document these changes and the new version number.