2023.02.14 Update wiki to include the detail of Radiam (simulation of point mutation).
ParasoR computes a variety of RNA secondary structure features for long RNA sequences even for human genome-level sequences by distributed computation using computer clusters.
Currently availabel features of ParasoR are
- Base pairing probability (bpp)
- Stem probability
- Accessibility (loop probability)
- Structure profiles (probability and motif sequence)
- Single-core mode: Minimum free energy (MFE) structure γ-centroid structure
- Multi-core mode: Maximum expected accuracy structure, which consists only the base pairs whose bpp is equal larger than 1/(1+γ))
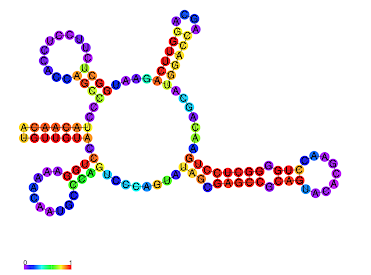
- γ-centroid structure with the color code of structure profiles.
- Color: Exterior (light green), Stem (red), Bulge (orange), Multibranch (green), Hairpin (violet), Internal (blue).
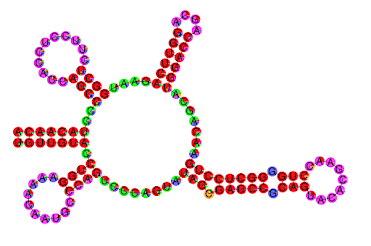
Additionally, ParasoR simulates structure arrangements caused by a single point mutation.
- C++11
We already tested ParasoR running with Apple LLVM version 6.0 and GCC 4.8.1.
git clone https://github.com/carushi/ParasoR
cd ParasoR
./configure
make
make install
Another way without git is downloading the directory directly from "Download ZIP" button.
As a default, a 'double' option is valid for the precision of floating point. This setting can be changed by editting the line in the makefile as below.
make VAR=LONG
# use long double.
make VAR=SHORT
# use float.
If you have a trouble about automake setting, please try a handmade makefile as shown below.
cd src
make -f _Makefile
or
autoreconf -ivf
A shell script 'check.sh' can be used for a test run. This script is exected by typing the commands as follows.
cd script/
sh check.sh
cat ../doc/pre.txt
# stem probability based on previous algorithm (Rfold model)
cat ../doc/stem.txt
# stem probability based on ParasoR algorithm
python test.py
# Output numerical error between the result of ParasoR with single core and multiple core
To see more samples, please visit our wiki.
- Kawaguchi R. et al. (2016) Parallel computation of genome-scale RNA secondary structure to detect structural constraints on human genome. BMC Bioinformatics, 17:203.
- Kiryu H. et al. (2008) Rfold: an exact algorithm for computing local base pairing probabilities. Bioinformatics, 24 (3), 367–373.
- Hamada M. et al. (2009) Prediction of RNA secondary structure using generalized centroid estimators. Bioinformatics, 25 (4), 465-473.
- Kiryu H. et al. (2011) A detailed investigation of accessibilities around target sites of siRNAs and miRNAs. Bioinformatics, 27 (13), 1789-97.
- Fukunaga T. et al. (2014) CapR: revealing structural specificities of RNA-binding protein target recognition using CLIP-seq data. Genome Biol., 15 (1), R16.
- Hamada M. et al. (2009) Prediction of RNA secondary structure using generalized centroid estimators. Bioinformatics, 25(4), 465–473.
- Gruber AR. et al. (2008) The Vienna RNA websuite. Nucleic Acids Res., 36 (Web Server issue), W70–W74.
- Turner DH. et al. (2010) NNDB: the nearest neighbour parameter database for predicting stability of nucleic acid secondary structure. Nucleic Acids Res., 38(Database issue), D280–D282.
- Andronescu M. et al. (2010) Computational approaches for RNA energy parameter estimation. RNA, 16(12), 2304–2318.