Haplotype marker based on 3,000 Rice Genomes (3K-RG) Project for genome-wide subpopulation ancestry inference.
The 3,000 Rice Genomes Project released the resequencing data of over 3,000 rice samples worldwide. 3K-RG population consists of two subspecies and multiple subpopulations. Within them, the four subpopulations with largest sample number are indica, tropical japonica, temperate japonica and aus.
Here I introduce a haplotype-based pipeline for subpopulation ancestry inference using 3K-RG as a background population.
NOTICE 1: This is a highly specialized pipeline and may be awkward for other uses. But some scripts in this pipeline may be usable for other situations.
NOTICE 2: The formulas and pictures in this page may not be displayed properly in some regions due to local Internet policies.
If you have any questions, suggestions or interests about this project, please feel free to contact: chenomics@163.com or zhuochen@fafu.edu.cn
20240614
A refined version of 3KRG-HAP pipeline is available in pipe_3KHAP.v20240614.zip. Take a try!
Pipeline of 3K-RG subpopulation marker construction and assignment:

Reference genome used in this pipeline is Nipponbare genome from: http://rice.uga.edu/
perl SNP_pruning.r2.pl --in 3K-RG.vcf --out 3K-RG.geno
This step yielded the 3K-SNP dataset. Named as 3K-SNP.geno in the following steps.
perl geno_to_binhap.pl --in 3K-RG.geno --out 3K-HAP.haplotype
This step yielded the 3K-RG haplotype file for each window. Named as 3K-HAP.haplotype
Assuming a population with m subpopulations, we defined a NAF-score (Normalized Allele Frequency score) of a certain subpopulation k for a certain haplotype in a 10-kb window following the equation below:
where n is the sample number of a subpopulation and a is the number of samples from a subpopulation that possess a certain haplotype in this window.
Need a tab-delimited list of 3K-RG sample names and subpopulation assignment. Named as 3K-RG.sample_list
perl haplotype_to_subtype_standard.pl 3K-HAP.haplotype 3K-RG.sample_list 3K-HAP.haplotype.NAF_score
This step yielded a NAF-scores for each subpopulation on each haplotype. Named as 3K-RG.haplotype.NAF_score.
Note: For the convenience of users, this file is provided in ./data/ and you can skip the first two steps.
Make a bed or interval file of SNPs in 3K-SNP dataset as required in GATK UnifiedGenotyper
Perform genotyping using GATK UnifiedGenotyper with these parameters: --L 3K-SNP.bed or --L 3K-SNP.intervals and --output_mode EMIT_ALL_SITES.
This step yielded VCF format genotype file of 3K-SNPs in custom population. Named as custom.vcf.
Note: 3K-SNP.bed is provided in ./data/
Make a varlist for haplotype construction with this command:
cat 3K-SNP.geno | grep -v "^#"| awk '{bin=int(($2-1)/10000);name=sprintf("%05d",bin);print $1"\t"$2"\t"$3"\t"$4"\t"$1"_"name}' > 3K-SNP.varlist
perl gatk_vcf_to_haplotype_with_varlist.pl --vcf custom.vcf --var 3K-SNP.varlist --out custom/haplotype/path/ --nohet
cat custom/haplotype/path/*.haplotype > custom.haplotype
Note: 3K-SNP.varlist is provided in ./data/
perl classify_sample_haplotype_score.pl custom.haplotype 3K-RG.haplotype.NAF_score outpath/
This step output NAF-score for each 10 kb window of each sample. Named as sample.NAF.
Then merge NAF-score for each 100 kb window.
perl scan_haplotype_stdratio.pl sample.NAF 10kb_window.bed sample.bin_NAF
perl dissect_rice_bin.pl sample.bin_NAF > sample.bin_NAF
Rscript draw_bin.rice.R chr.len sample.bin_NAF sample.bin_NAF.pdf
An example, genome-wide subpopulation ancestry inference of an elite Chinese rice cultivar DHX2 ("稻花香2号"):
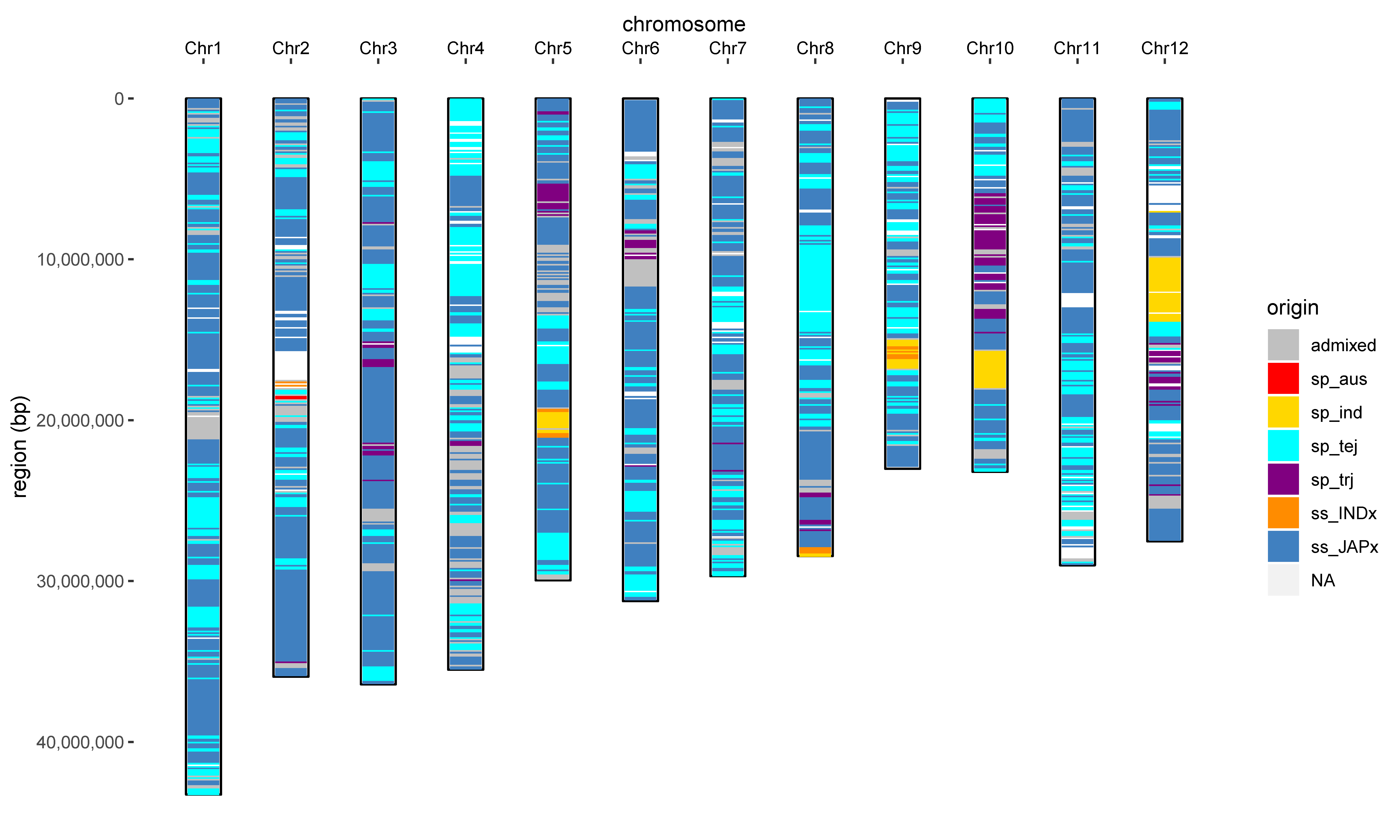
NGS data of this DHX2 sample was obtained from Zhao et al., Nat.Genet. 2018.
Li JY, Wang J, Zeigler RS. The 3,000 rice genomes project: new opportunities and challenges for future rice research. Gigascience. 2014;3:8. Published 2014 May 28. doi:10.1186/2047-217X-3-8
3,000 rice genomes project. The 3,000 rice genomes project. Gigascience. 2014;3:7. Published 2014 May 28. doi:10.1186/2047-217X-3-7
Wang W, Mauleon R, Hu Z, et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature. 2018;557(7703):43–49. doi:10.1038/s41586-018-0063-9
Zhao Q, Feng Q, Lu H, et al. Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice [published correction appears in Nat Genet. 2018 Aug;50(8):1196]. Nat Genet. 2018;50(2):278–284. doi:10.1038/s41588-018-0041-z
Zhuo Chen, Xiuxiu Li, Hongwei Lu, Qiang Gao, Huilong Du, Hua Peng, Peng Qin, Chengzhi Liang. Genomic atlases of introgression and differentiation reveal breeding footprints in Chinese cultivated rice. Journal of Genetics and Genomics, 2020, ISSN 1673-8527, https://doi.org/10.1016/j.jgg.2020.10.006.