Warning: This package is still under constrution.


ENPMDA is a parallel analysis package for ensemble simulations powered by MDAnalysis.
It stores metadata in pandas.DataFrame
and distributes computation jobs in dask.DataFrame
so that the parallel analysis can be performed
not only for one single trajectory
but also across simulations and analyses.
It can be used as an initial inspection of the raw trajectories as well as a framework for extracting features from final production simulations for further e.g. machine learning and markov state modeling. It automatically fixes the PBC issue, and align and center the protein inside the simulation box. It also works for multimeric proteins!
The framework is intended to be adaptable by being able to simply wrapping MDAnalysis analysis functions without worrying about the parallel machinery behind.
- Free software: GNU General Public License v3
- Documentation: https://ENPMDA.readthedocs.io.
- Parallel analysis for ensemble simulations.
- Dataframe for storing and accessing results.
- dask-based task scheduler, suitable for both workstations and clusters.
- Expandable analysis library powered by MDAnalysis.
from ENPMDA import MDDataFrame
from ENPMDA.preprocessing import TrajectoryEnsemble
from ENPMDA.analysis import get_backbonetorsion, rmsd_to_init
# construct trajectory ensemble
traj_ensemble = TrajectoryEnsemble(
ensemble_name='ensemble',
topology_list=ensemble_top_list,
trajectory_list=ensemble_traj_list
)
traj_ensemble.load_ensemble()
# initilize dataframe and add trajectory ensemble
md_dataframe = MDDataFrame(dataframe_name='dataframe')
md_dataframe.add_traj_ensemble(traj_ensemble, npartitions=16)
# add analyses
md_dataframe.add_analysis(get_backbonetorsion)
md_dataframe.add_analysis(rmsd_to_init)
# save dataframe
md_dataframe.save('results')
# retrieve feature
feature_dataframe = md_dataframe.get_feature([
'torsion',
'rmsd_to_init'
])
# plot analysis results
import seaborn as sns
sns.barplot(data=feature_dataframe,
x='system',
y='rmsd_to_init')
sns.lineplot(data=feature_dataframe,
x='traj_time',
y='0_phi_cos',
hue='system')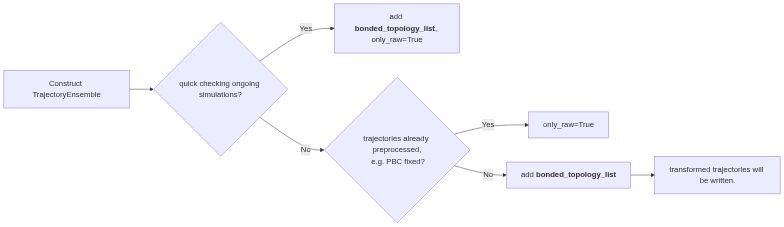

For a system of 250,000 atoms (1500 protein residues), the total time for analyzing 220,000 frames of
- RMSD to initial frame
- Pore hydration
- All protein torsion angle
- All C-alpha positions
- 15,000 pair-wise distances
is 10 minutes using 5 nodes in Dardel (640 cores).

- option to add more than one ensemble
- more analysis functions.
- unit testing
- benchmarking
- documentation
- add functions to cancel running tasks
- MDAnaysis: https://www.mdanalysis.org/
- pmda: https://github.com/mdAnalysis/pmda
- dask: https://dask.org/
This package was created with Cookiecutter and the audreyr/cookiecutter-pypackage project template.