This is a repository to reproduce our KIR genotyping method (Sakaue et al.)
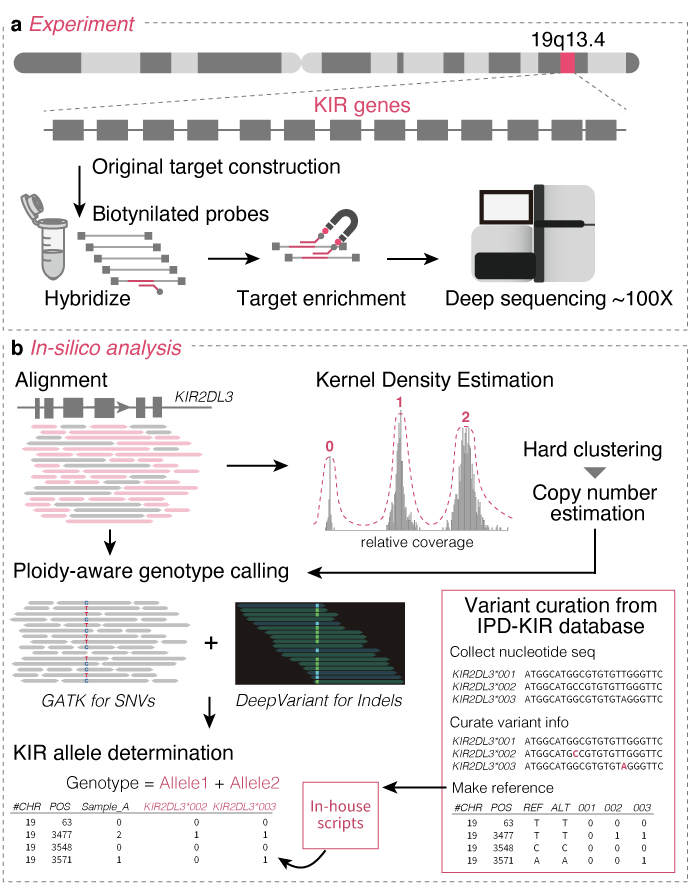
There are three levels of variations in KIR genes, and we address determining these variations using target sequencing data and our bioinformatics pipeline.

01_Map_to_KIRreference.sh is our mapping script using picard and bwa mem.
| Var | Descriptions |
|---|---|
| SAMPLEPATH | Path to the fastq files |
| ID | Sample ID to be used in bam files |
| BAMPATH | Path to the output bam files |
| COVERAGE | Path to the depth of coverage (to be used to determine the copy number) |
02_determine_copy_number.py is a script to determine the copy number of each KIR genes using KIR3DL3 as a reference, using Kernel Density Estimation.
| Var | Descriptions |
|---|---|
| input_csv | A csv file describing each KIR gene (row)'s mean depth per sample (column) |
| output_ploidy_file | Output file indicating an estimated ploidy per sample per gene (per each line) |
Using the bam file and KIR copy number obtained in the previous steps, we can call genotypes per sample per gene by using HaplotypeCaller with 03_HC.by_ploidy.sh, and jointly genoype across samples by 04_JG.by_ploidy.sh.
| Var | Descriptions |
|---|---|
| GENE | A gene to call genotype |
| PLOIDY | Number indicating the ploidy (copy number) per gene per sample (integer) |
| GVCFPATH | Path to output GVCFs |
We use specific indels to determine KIR alleles. For that purpose, 05_run_deepvariant.sh can do genotyping using DeepVariant.
| Var | Descriptions |
|---|---|
| DeepVariantPATH | Path to output DeepVariant VCFs and GVCFs |
06_genotype_call_with_ploidy.py can determine possible KIR allele combination(s) per gene per sample.
| Var | Descriptions |
|---|---|
| per_sample_vcf_file | output GVCFs/DeepVariant GVCFs (Note to split the multiallelic variants) |
| output_allele_file | output file for possible KIR allele combination(s) |
| dosage_file | interim file for collecting variant information |
| reference_file | interim file for collecting reference variant information |
Questions to: ssakaue_at_broadinstitute.org