Submission: phylogram #212
Comments
|
thanks for your submission @shaunpwilkinson - will get to editor checks tomorrow morning |
Editor checks:
Editor commentsThanks for your submission @shaunpwilkinson !!
── GP phylogram ───────────
It is good practice to
✖ write unit tests for all functions, and all package code in general. 82% of code lines are covered by test cases.
R/ladder.R:33:NA
R/ladder.R:34:NA
R/ladder.R:35:NA
R/ladder.R:36:NA
R/ladder.R:37:NA
... and 63 more lines
✖ not use "Depends" in DESCRIPTION, as it can cause name clashes, and poor interaction with other packages. Use "Imports" instead.
✖ avoid long code lines, it is bad for readability. Also, many people prefer editor windows that are about 80 characters wide. Try make your lines
shorter than 80 characters
R/prune.R:66:1
R/prune.R:96:1
R/read.R:74:1
R/read.R:151:1
R/write.R:32:1
... and 2 more lines
✖ avoid sapply(), it is not type safe. It might return a vector, or a list, depending on the input data. Consider using vapply() instead.
R/ladder.R:35:21
R/prune.R:43:21
R/read.R:113:25
R/read.R:114:26
R/read.R:158:21
... and 3 more lines
✖ not import packages as a whole, as this can cause name clashes between the imported packages. Instead, import only the specific functions you need.
✖ not use exportPattern in NAMESPACE. It can lead to exporting functions unintendedly. Instead, export functions that constitute the external API of
your package.Seeking reviewers now 🕐 Reviewers: @benjward @wcornwell |
|
Thanks very much @sckott I will get onto those good-practice edits asap! S |
|
sorry for delay, still trying to find 2 reviewers 🕐 |
|
Reviewers assigned: Reviewers: @benjward @wcornwell |
Package ReviewPlease check off boxes as applicable, and elaborate in comments below. Your review is not limited to these topics, as described in the reviewer guide
DocumentationThe package includes all the following forms of documentation:
Functionality
Final approval (post-review)
Estimated hours spent reviewing: 3 --- Review CommentsThanks for this package. I didn't know anything really about the dendrogram class in R before looking into this. And it does look like a useful thing for the R-phylo-universe to have ways to go back and forth between the edge matrix style and the nested list style representation. There might be interesting applications in between taxonomy and phylogenies, and it's always good to have a full suite of tools to efficiently translate between data structures. As taxonomy moves to Non-linnaean data sets, the nested list structure might become increasingly useful. I thought I would get to this quickly, and just do some of my normal research with this new tool and see how it goes. The tree that we have been using for the last several years is available (here)[https://datadryad.org//resource/doi:10.5061/dryad.63q27]. It's a ~30,000 tip tree for angiosperms, and typically we cut it down to a subset for whatever the task at hand is, but when I tried to load it using evolve.tree <- function(size){
object.size(diversitree::tree.bd(pars=c(1,0),max.taxa=size))
}
evolve.dendro <- function(size){
object.size(phylogram::as.dendrogram(diversitree::tree.bd(pars=c(1,0),max.taxa=size)))
}
x <- seq(2, 200, 10)
df <- data.frame(ape.size=purrr::map_dbl(x,evolve.tree),
dendro.size=purrr::map_dbl(x,evolve.dendro))
library(ggplot2)
ggplot(df, aes(x=ape.size,y=dendro.size)) + geom_point()+theme_bw()It turns out Then I checked read in speeds: read_tree_dendro<-function(size){
ape::write.tree(diversitree::tree.bd(pars=c(1,0),max.taxa=size),"twoh.tre")
system.time({phylogram::read.dendrogram("twoh.tre")})[3]
}
read_tree_ape <- function(size){
ape::write.tree(diversitree::tree.bd(pars=c(1,0),max.taxa=size),"twoh.tre")
system.time({ape::read.tree("twoh.tre")})[3]
}
set.seed(42)
x <- seq(100,3000,150)
df <- data.frame(phylo_size=x,
ape.speed=purrr::map_dbl(x, read_tree_ape),
dendro.speed=purrr::map_dbl(x, read_tree_dendro))
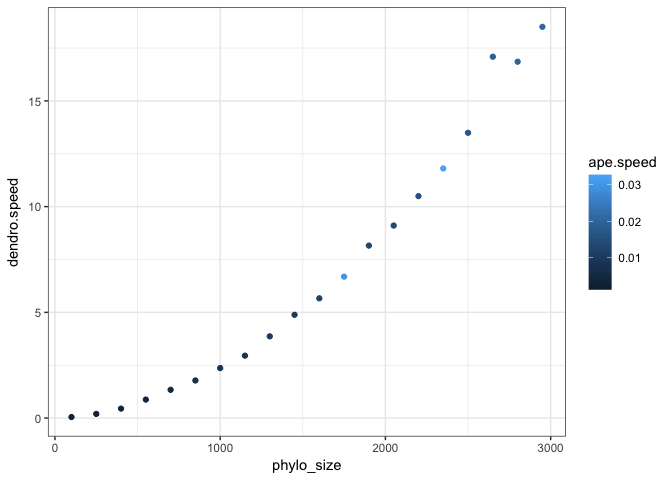
ggplot(df, aes(x=ape.speed,y=dendro.speed,col=phylo_size)) + geom_point() + theme_bw()ggplot(df, aes(x=phylo_size,y=dendro.speed,col=ape.speed)) + geom_point() + theme_bw()So I guess there is something non-linear about the performance of I tried to read the Phylogenies are getting bigger all the time: there was a 300,000 tip tree for plants published this week. And there are some giant bacterial trees out also. So for the functionality of this package, designing and optimising the code for at least medium sized trees is pretty crucial. other notes:
library(phylogram)
newick <- "(A:0.1,B:0.2,C:0.3),D:0.4:0.5);"
x <- read.dendrogram(text = newick)xand also newick <- "(A:0.1,(B:0.2,)C:0.3,D:0.4);"
x <- read.dendrogram(text = newick)x |
figures from above
|


|
thanks for your review @wcornwell ! |
|
Thank you very much for your review @wcornwell I'll make it a priority to optimize the parser to better handle those bigger trees and add more informative error messages for when the tree parse fails. Will aim to get these and your other suggestions seen to in the next week or two. Thanks again for the feedback! |
|
Hi @sckott, I have now made the changes suggested by @wcornwell, including a major overhaul of the read.dendrogram and as.dendrogram.phylo functions which are now much more efficient. Just wondering am I okay to push the changes to GitHub or would you prefer I leave the repo alone until the other review comes in? |
|
@shaunpwilkinson great, thanks. Can you please include a response here to @wcornwell suggestions, it doesn't have to be exactly point by point, but a summary at least. We don't really have rules about how changes are made with respect to the two different reviewers, but I think it's a good idea to wait for the 2nd review to come in. |
|
Thanks @sckott, will do, and no problem to hold off pushing the changes until the second review is finished. |
Package Review
DocumentationThe package includes all the following forms of documentation:
Functionality
Final approval (post-review)
Estimated hours spent reviewing: ~3 hours. Review CommentsThis package will certainly be useful to convert the specific tree classes in Checking, installation, and testing👍 The coverage of the source code is good: phylogram Coverage: 85.18%
R/read.R: 76.26%
R/prune.R: 79.73%
R/write.R: 94.74%
R/utils.R: 94.87%
R/asdendrogram.R: 100.00%
R/asphylo.R: 100.00%
R/ladder.R: 100.00%SuggestionCoverage could be improved slightly in read.R and prune.R. In particular, it would be good to add to the test suite some cases testing that UseageGoing through the examples in the vignettes was simple and all examples ran for me. I won't comment on performance as that has already been addressed in previous SuggestionsIn the vignette, I think following the instructions on manipulating trees directly i.e.: Is important to demonstrate how the trees can be built up from a vanilla R linked I think that immediately following this, the vignette could demonstrate how to I think that if this was added it would make users much more comfortable with manipulating trees in a much more semantic way. In the future - and this is not limited to this package, it would be cool to see ConcludingThis is a nice and simple R package, which adds compatibility between a few different Thank you for this package 😄 |
|
thanks for your review @benjward ! @shaunpwilkinson all reviews in ✅ |
|
thanks! |
|
Hi @sckott, I have now pushed the changes to GitHub and will release a new version of the package on CRAN shortly. We are pleased that the reviewers found the package useful, and are very happy with the improvements made to the package and vignette. Our responses to the reviewer comments/suggestions are listed below. @wcornwell and @benjward thank you both so much for the extremely helpful reviews and advice on improving the package. The detailed write-ups you provided were an immense help for optimizing the package functions and making vignette more user friendly. We carried out a fairly major overhaul on the package (see commit # Review 1 - @wcornwell Comment: There is something non-linear about the performance of Response: The previous version of Comment: I'm sure there are some use cases for comparative methods where a nested list will be more useful than an edge matrix. Be nice to have a worked example in the vignette. Response: Great idea, we have now included an example in the vignette on how to convert between phylo and dendrogram objects to create tanglegrams using the dendextend package. This seems to be a commonly encountered issue (see threads here, here , and here ), so I think this constitutes a really important addition to the vignette. Comment: It would also be worth comparing the performance of Response: Yes we agree, and have added a couple of sentences to the vignette as follows: Leaf nodes and internal branching nodes can be removed using the function Comment: Reading in problematic newick strings is kind of a nightmare because there are so many flavors of errors, but there are probably some cases where read.dendrogram returns a warning, when it really should give an informative error message rather than the current behavior which is to return a warning and a garbled tree. Response: This was another factor in our decision to decommission the parser and instead focus on extending ape’s (much better) Review 2- @benjward Comment: Coverage could be improved slightly in read.R and prune.R. In particular, it would be good to add to the test suite some cases testing that errors are indeed thrown, when they are expected to be thrown - by giving package functions deliberately incorrect input, and using testthat to catch the errors with Response: We have now expanded the unit tests to cover 95% of the code (including 100% for read.R and 96% for prune.R) and have also added an Comment: In the vignette, I think following the instructions on manipulating trees … is important to demonstrate how the trees can be built up from a vanilla R linked data structure, but this style becomes difficult to read quite quickly for non-trivial trees. I think that immediately following this, the vignette could demonstrate how to build the same tree, but by using functions that define a set of verbs common to phylogentics (like Response: This is a really good point, and would certainly help users get started with some basic manipulation of dendrogram objects. We have now rearranged the vignette to demonstrate the ground-up tree building towards the end (after the basic reading, writing and conversion functions) and added a new example showing how to build and manipulate the same basic tree object with the package functions. We think this adds a valuable contribution to the tutorial. Comment: In the future - and this is not limited to this package, it would be cool to see Response: Yes we agree, and did consider adding a new method to ladderize for dendrogram objects, but unfortunately there was a clash with one of the functions in the dendextend package and so we decided to keep them separate. Of course we are happy to take advice on this! |
|
thanks @shaunpwilkinson for the responses to reviewers. @wcornwell @benjward are you happy with the responses to your comments? |
|
@sckott Absolutely 👍 |
|
thx @benjward |
|
@wcornwell are you happy with the changes made? |
|
sounds good! @sckott |
|
@shaunpwilkinson both reviewers are on board with changes. I'll do a quick peek ... |
|
@shaunpwilkinson one question:
|
|
Hi @sckott I spent ages trying to get the NAMESPACE right using roxygen tags and |
|
Nah, seems good as is. Approved! Thanks again for your submission @shaunpwilkinson
|
|
That's great news - thanks so much @sckott ! I'll get to those tasks today. Quick question - I'm planning to submit the paper to JOSS and wondering if it will be a problem if the |
|
@shaunpwilkinson good question about JOSS, i don't know. @noamross do you know answer to the above? whether JOSS cares if the paper is also somewhere else in another form? |
|
Hi @sckott I can't see an invite to join a team within the ropensci organization - any chance you could point me in the direction of where I should be looking? Perhaps I'm just being blind. |
|
sorry about that, you should have gotten it now |
|
No worries, almost there, I've managed to join the organization but when I go to transfer the repo I get the error message |
|
woops, sorry. Github recently changed how permissions work for transferring repos to organizations. can you add me as admin to your repo - then i'll transfer to ropensci - then you can take it from there? |
|
No prob, I've just added you as a collaborator - I'm assuming that's the same thing.. |
|
thanks! not quite.
i need to be able to get to Settings tab, so i need admin access |

|
Hmm, I don't seem to have that option, I just had a look at the GitHub help pages (https://help.github.com/articles/permission-levels-for-a-user-account-repository/#collaborator-access-on-a-repository-owned-by-a-user-account) and it looks like there are only two permission levels, owner and collaborator, so I have just transferred the ownership to you |
|
thanks! and just moved to ropensci, and you have admin access on it, let me know if you don't have access |
|
Are you interested in doing a blog post for our blog https://ropensci.org/blog/ ? either a short-form intro to it (https://ropensci.org/technotes/) or long-form post with more narrative about its development (https://ropensci.org/blog/). If so, we'll have our community manager @stefaniebutland get in touch with you on that |
|
Hi @sckott and @stefaniebutland, yes I'd be more than happy to, I'll write up a technote asap. Also just FYI the latest version of the package is on CRAN now, and JOSS paper is online too (http://joss.theoj.org/papers/10.21105/joss.00790) |
|
Glad to hear you'll contribute a technote @shaunpwilkinson. Will tweet it from @ropensci to get more eyes on your work. Here's info on how to submit via pull request https://github.com/ropensci/roweb2#contributing-a-blog-post. Difference in YAML:
Since Tech Notes can be published any time, you can choose the date, but please submit about a week in advance so we can review. Happy to answer any questions that come up. |
|
Forgot to mention, please add these tags to YAML (among others)
And be sure that the tech note gives an example of a cool thing you can do with the package, rather than reiterating the README for example. Thank you! |
Summary
The phylogram R package is a tool for for developing phylogenetic trees as deeply-nested lists known as "dendrogram" objects. It provides functions for importing and exporting trees in parenthetic text format, as well as several tools for command-line tree manipulation and conversion between "dendrogram" and "phylo" class objects.
https://github.com/shaunpwilkinson/phylogram
[e.g., "data extraction, because the package parses a scientific data file format"]
Data extraction, the package contains a text parser for trees in the Newick parenthetic text format, and functions for converting between dendrogram and phylo object types.
Biologists and bioinformaticists wishing to access the many analytical and tree-visualization functions available for dendrogram objects in R.
yours differ or meet our criteria for best-in-category?
There are other packages that can parse Newick strings to create phylo objects (e.g. ape, ggtree) but none that we are aware of that enable two-way conversion between phylo and dendrogram objects.
Requirements
Confirm each of the following by checking the box. This package:
Publication options
paper.mdmatching JOSS's requirements with a high-level description in the package root or ininst/.Detail
R CMD check(ordevtools::check()) succeed? Paste and describe any errors or warnings:No errors or warnings.
Does the package conform to rOpenSci packaging guidelines? Please describe any exceptions:
If this is a resubmission following rejection, please explain the change in circumstances:
If possible, please provide recommendations of reviewers - those with experience with similar packages and/or likely users of your package - and their GitHub user names:
Tal Galili (talgalili)
Zuguang Gu (jokergoo)
Michael Krabbe Borregaard (mkborregaard)
Guangchuang Yu (GuangchuangYu)
The text was updated successfully, but these errors were encountered: