Home
This wiki will walk through the installation and use of DigDriver to identify cancer driver elements anywhere in the genome in a cancer cohort of interest. Tutorials will demonstrate how to run analyses from Sherman et al. 2021.
Simply progress through the pages in the sidebar in order to install DigDriver, download necessary data files, and begin testing for positive selection.
The purpose of DigDriver is to test for positive selection anywhere in the genome in any cohort of sequenced tumor samples. Positive selection manifests as an increase in the number of mutations observed in the tumor cohort compared to the number expected under the neutral mutation rates that affect all cells in the body. The key component of DigDriver is the mutation map model. These models contain 1) cancer-specific mean and variance estimates of the mutation rate in kilobase-scale regions tiled across the genome and 2) estimates of the nucleotide context biases of cancer-specific mutational processes. Together, these two components enable any set of mutations in the genome to be tested for positive selection in milliseconds in a set of somatic mutations provided by the user. The figure below provides a graphical overview of the DigDriver method. The method is described in detail in our paper Sherman et al. 2021 (LINK).
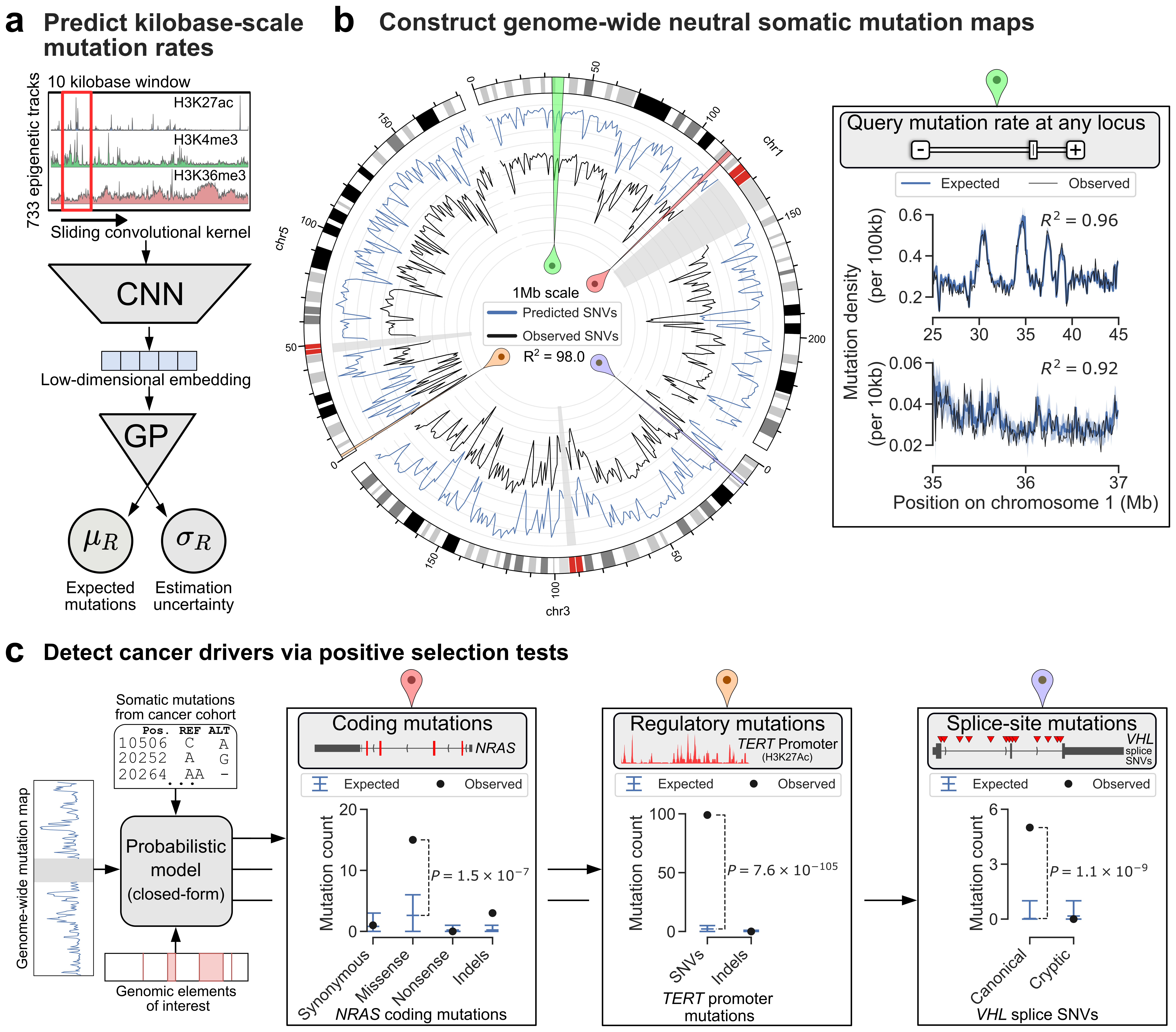
DigDriver offers three primary utilities for identifying positive selection:
The genic driver model searches for burdens of mutations in the canonical transcript of 19,210 genes, stratifying by predicted coding impact (synonymous, missense, nonsense, etc.). Details are provided here (LINK).
This model calculates mutational burden across arbitrary sets of contiguous or noncontiguous genomic intervals. Details are provided here (LINK).
This model calculates mutational burden across any arbitrary set of SNVs. Details are provided here (LINK).>