##Getting an account
To request a Beocat account go to https://account.beocat.cis.ksu.edu/user.
##Log onto Beocat
###On Macs
Find and open your shell. On Macs the Terminal is in Applications/Utilities/Terminal.
From your command prompt log on to Beocat by typing (enter your EID in the place of "EID"). Next type your password (the same one you use to log onto KSOL). When you type your password you will not see any text on the screen (this protects your privacy). The first time you login you will be asked if you want to continue. Type "yes":
ssh EID@beocat.cis.ksu.edu
###On PCs
On PCs download and install putty.exe from http://www.chiark.greenend.org.uk/~sgtatham/putty/download.html.
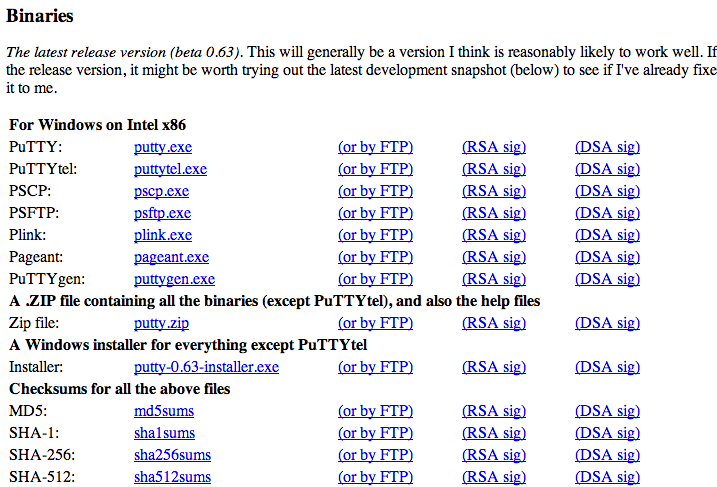
Open PuTTY and type "EID@beocat.cis.ksu.edu" (enter your EID in the place of "EID") into the "Host Name (or IP address) box.:
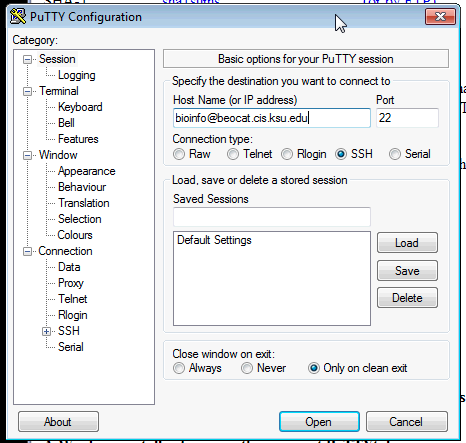
Click yes to add the Key to PuTTY's cache.
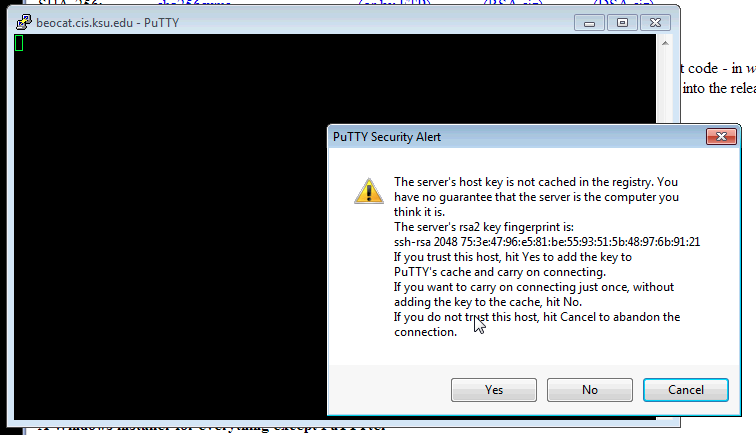
Next type your password (the same one you use to log onto KSOL) into the terminal. When you type your password you will not see any text on the screen (this protects your privacy).
##Basic Unix commands
A few simple cammands can be very useful for using Beocat. These include:
###tab to autocomplete
If you are typing a path into the commandline you can start typing and if the start of the path is unique the terminal will finish the path for you. Otherwise it will complete part of the path and print all possible ways to finish the path on your screen.
##Up and down arrows
Use the up and down arrows on your keyboard to find previous commands. Press enter to execute them.
##cd
Change directories with "cd".
cd [directory]
Example:
cd /homes/bioinfo/bioinfo_software
##ls
List the contents of the current directory by typing "ls".
ls [directory]
Example:
ls /homes/bioinfo/bioinfo_software
##less,head,tail
Use "less" to read a file. Use up and down arrows to read the file. Type "q" to exit.
less [filename]
Example:
less /homes/bioinfo/pipeline_datasets/RNA-SeqAlign/cell_line_reads_DE.txt
Use "head" to read the first few lines of a file.
head /homes/bioinfo/pipeline_datasets/RNA-SeqAlign/MB468_paired-end_RNA-seq_subsampled_2.fastq
Use "tail" to read the last few lines of a file.
tail [filename]
Example:
tail /homes/bioinfo/pipeline_datasets/RNA-SeqAlign/MB468_paired-end_RNA-seq_subsampled_2.fastq
If you would like a quick primer on basic linux commands try these 10 minute lessons from Software Carpentry http://software-carpentry.org/v4/shell/index.html. For Beocat basics see http://support.cis.ksu.edu/BeocatDocs/GettingStarted.