This subworkflow prepares tracks to represent external annotation. NCBI is usually the source but any appropriate fasta can be mapped to the genome being reported. Fasta files containing known RNA transcripts, coding sequences, genes and proteins as amino acids can be mapped using the Miniprot and Minimap2 tools, with bam to bed format conversion where needed. Unlike the TreeVal subworkflow it replaces, tabix indexing is delegated to the integrated GMOD JBrowse2 tool downstream in the main TreeValGal workflow, and the tracks are integrated into the interactive browser output, ready to view in Galaxy, as shown in this example for the VGP Calypte anna (Hummingbird) assembly, where one of the NCBI protein track features has been selected, and the annotation for that feature has popped up on the right side of the browser window.


This Galaxy workflow canvas is the entire workflow from a user's perspective. The GUI used to build and update workflows, exposes all the inputs, logic, tools and data flows, allowing tool and other settings to be accessed and configured with the Galaxy input forms. All Galaxy tools are interoperable in workflows, without ever requiring any user supplied code for data flows between them or for setting parameters. Data flows are configured by drawing the connections between outputs and inputs, on the canvas with a mouse pointing device. Tool settings are adjusted using the usual tool forms, without the workflow developer needing to learn anything about their internal code or configuration.
This is in contrast to the original Nextflow TreeVal subworkflow described below, requiring 284 lines of DDL to be written by the workflow developer, to correctly pass data and parameters suitable for the modules used in the subworkflow. The developer must examine those modules to learn what parameter and data names and formats must be passed. Nextflow does not provide any automated interface between modules.
Of the 23 elements in the Galaxy workflow, 7 are tool steps, 5 are run-time data selections and 11 are specialised workflow control logic elements. They are usually not needed, but are configured to allow optional inputs to tools that do not allow them. They are used here for VGP species where little or no external annotation is available. Missing annotation inputs can be left off for any run, without creating any empty browser tracks.
Specialised workflow inputs and logic elements are added to the usual tool menu during workflow GUI editing sessions. Tool menu items can be selected and dropped onto the canvas, ready to be configured.
The pick component is used here to implement optional workflow inputs for tools that do not natively support them. Execution of those tool steps is prevented tools to avoid failure if there is no input available, using a
boolean GUI input yes/no toggle for the user to click at run-time. While they look complicated, each requires only a few mouse clicks, and some text labels, to be ready for use.
When an edited workflow is saved, all metadata and other information required to configure the workflow inputs and to run all the steps, is automatically generated in the form of a shareable JSON document,
for download and storage on the server, that never needs manual editing.
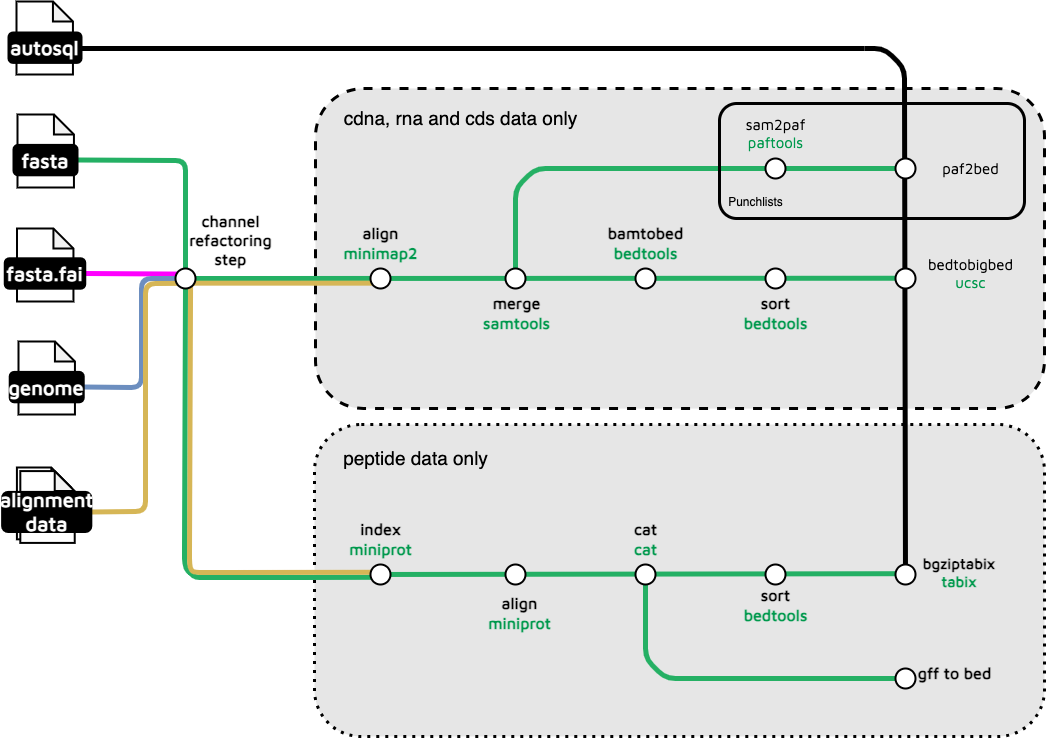
The NextFlow code to implement this flow chart requires a total of 284 lines of custom written DDL:
../subworkflows/local/nuc_alignments.nf 16 41 121
../subworkflows/local/gene_alignment.nf 14 24 84
../subworkflows/local/pep_alignments.nf 14 35 79
This count excludes all the other DDL in dependencies called by these 3 custom written files, assuming dependencies required no additional coding for their use in this subworkflow.
treeval_upload/
*.gff.gz: Zipped .gff for each species with peptide data.
*.gff.gz.tbi: TBI index file of each zipped .gff.
*_cdna.bigBed: BigBed file for each species with complementary DNA data.
*_cds.bigBed: BigBed file for each species with nuclear DNA data.
*_rna.bigBed: BigBed file for each species with nRNA data.
treeval_upload/punchlists/
*_pep_punchlist.bed: Punchlist for peptide track.
*_cdna_punchlist.bed: Punchlist for cdna track.
*_cds_punchlist.bed: Punchlist for cds track.
*_rna_punchlist.bed: Punchlist for rna track.
dot_genome // Channel [ val(meta), path(file) ]
reference_tuple // Channel [ val(meta), path(file) ]
reference_index // Channel [ val(meta), path(file) ]
max_scaff_size // Channel val(size of largest scaffold in bp)
assembly_classT // Channel val(clade_id)
alignment_datadir // Channel val(geneset_dir)
alignment_genesets // Channel val(geneset_id)
alignment_common // Channel val(common_name) // Not yet in use
intron_size // Channel val(50k)
as_files // Channel [ val(meta), path(file) ]
main:
The subworkflow DDL has function calls explained below.
// This subworkflow takes an input fasta sequence and csv style list of organisms to return
// bigbed files containing alignment data between the input fasta and csv style organism names.
// Input - Assembled genomic fasta file
// Output - A BigBed file per datatype per organism entered via csv style in the yaml.
//
// SUBWORKFLOW IMPORT BLOCK
//
include { PEP_ALIGNMENTS } from './pep_alignments'
include { NUC_ALIGNMENTS as GEN_ALIGNMENTS } from './nuc_alignments'
include { NUC_ALIGNMENTS as RNA_ALIGNMENTS } from './nuc_alignments'
include { NUC_ALIGNMENTS as CDS_ALIGNMENTS } from './nuc_alignments'
workflow GENE_ALIGNMENT {
take:
dot_genome // Channel [ val(meta), path(file) ]
reference_tuple // Channel [ val(meta), path(file) ]
reference_index // Channel [ val(meta), path(file) ]
max_scaff_size // Channel val(size of largest scaffold in bp)
assembly_classT // Channel val(clade_id)
alignment_datadir // Channel val(geneset_dir)
alignment_genesets // Channel val(geneset_id)
alignment_common // Channel val(common_name) // Not yet in use
intron_size // Channel val(50k)
as_files // Channel [ val(meta), path(file) ]
main:
ch_versions = Channel.empty()
//
// LOGIC: TAKES A SINGLE LIKE CSV STRING AND CONVERTS TO LIST OF VALUES
// LIST IS MERGED WITH DATA_DIRECTORY AND ORGANISM_CLASS
//
ch_data = alignment_genesets
.splitCsv()
.flatten()
//
// LOGIC: COMBINE CH_DATA WITH ALIGNMENT_DIR AND ASSEMBLY_CLASS
// CONVERTS THESE VALUES INTO A PATH AND DOWNLOADS IT, THEN TURNS IT TO A TUPLE OF
// [ [ META.ID, META.TYPE, META.ORG ], GENE_ALIGNMENT_FILE ]
// DATA IS THEN BRANCHED BASED ON META.TYPE TO THE APPROPRIATE
// SUBWORKFLOW
//
ch_data
.combine( alignment_datadir )
.combine( assembly_classT )
.map {
ch_org, data_dir, classT ->
file("${data_dir}${classT}/csv_data/${ch_org}-data.csv")
}
.splitCsv( header: true, sep:',')
.map( row ->
tuple([ org: row.org,
type: row.type,
id: row.data_file.split('/')[-1].split('.MOD.')[0]
],
file(row.data_file)
))
.branch {
pep: it[0].type == 'pep'
gen: it[0].type == 'cdna'
rna: it[0].type == 'rna'
cds: it[0].type == 'cds'
}
.set {ch_alignment_data}
pep_files = ch_alignment_data.pep.collect()
gen_files = ch_alignment_data.gen.collect()
rna_files = ch_alignment_data.rna.collect()
cds_files = ch_alignment_data.cds.collect()
//
// SUBWORKFLOW: GENERATES GENE ALIGNMENTS FOR PEPTIDE DATA, EMITS GFF AND TBI
//
PEP_ALIGNMENTS ( reference_tuple,
pep_files,
max_scaff_size
)
ch_versions = ch_versions.mix(PEP_ALIGNMENTS.out.versions)
//
// SUBWORKFLOW: GENERATES GENE ALIGNMENTS FOR RNA, NUCLEAR AND COMPLEMENT_DNA DATA, EMITS BIGBED
//
GEN_ALIGNMENTS ( reference_tuple,
reference_index,
gen_files,
dot_genome,
intron_size
)
ch_versions = ch_versions.mix( GEN_ALIGNMENTS.out.versions )
CDS_ALIGNMENTS ( reference_tuple,
reference_index,
cds_files,
dot_genome,
intron_size
)
ch_versions = ch_versions.mix( CDS_ALIGNMENTS.out.versions )
RNA_ALIGNMENTS ( reference_tuple,
reference_index,
rna_files,
dot_genome,
intron_size
)
ch_versions = ch_versions.mix( RNA_ALIGNMENTS.out.versions )
emit:
pep_gff = PEP_ALIGNMENTS.out.tbi_gff
gff_file = PEP_ALIGNMENTS.out.gff_file
gen_bb_files = GEN_ALIGNMENTS.out.nuc_alignment
rna_bb_files = RNA_ALIGNMENTS.out.nuc_alignment
cds_bb_files = CDS_ALIGNMENTS.out.nuc_alignment
versions = ch_versions.ifEmpty(null)
The .branch() DDL _appears to allow optional data dependent alignments for peptides, dna, rna and cds inputs. These in turn involve PEP_ALIGNMENTS and NUC_ALIGNMENTS subworkflows.
They are complex and have their own decompositions at nuc_alignments and pep_alignments.
They in turn involve miniprot-align available from the IUC in the Toolshed, and PAF2BED and PAFTOOLS that appear to be part of minimap so perhaps supplied from the suite in the Toolshed, plus things already in the Toolshed like samtools_faidx.
include { MINIMAP2_ALIGN } from '../../modules/nf-core/minimap2/align/main'
include { SAMTOOLS_MERGE } from '../../modules/nf-core/samtools/merge/main'
include { SAMTOOLS_FAIDX } from '../../modules/nf-core/samtools/faidx/main'
include { BEDTOOLS_SORT } from '../../modules/nf-core/bedtools/sort/main'
include { BEDTOOLS_BAMTOBED } from '../../modules/nf-core/bedtools/bamtobed/main'
include { UCSC_BEDTOBIGBED } from '../../modules/nf-core/ucsc/bedtobigbed/main'
include { PAFTOOLS_SAM2PAF } from '../../modules/nf-core/paftools/sam2paf/main'
include { PAF2BED } from '../../modules/local/paf_to_bed'
The other steps needed for this subworkflow are:
PAF2BED is a local module that calls the following command line :
paf_to_bed.sh ${file} ${prefix}.bed
If this is only needed for JBrowse as noted in the code, then it can be ignored because JB2 can display paf.
That bash script is in /treeval/bin and is an exotic awk exercise.
#!/bin/bash
# paf_to_bed12.sh
# -------------------
# A shell script to convert a
# paf into bed format for use
# in JBrowse
# -------------------
# Author = yy5
version='1.0.0'
if [ $1 == '-v'];
then
echo "$version"
else
cat $1 | awk 'BEGIN{FS="\t";}{a[$1]++;if(a[$1]==2)print v[$1] ORS $0;if(a[$1]>2)print;v[$1]=$0;}' | awk '$(NF+1) = ($10/$11)*100' | awk '$(NF+1) = ($10/$2)*100' | awk -vOFS='\t' '{print $6,$8,$9,$1,$2,$10,$(NF-1),$NF}' > $2
fi
PAFTOOLS_SAM2PAF uses samtools and perhaps another minimap suite script ? Looks like a new tool is needed for that undocumented jar.
samtools view -h ${bam} | paftools.js sam2paf - > ${prefix}.paf