By shlee

We provide example data and example commands for performing local realignment around small insertions and deletions (indels) against a reference. The resulting BAM reduces false positive SNPs and represents indels parsimoniously. First we use RealignerTargetCreator to identify and create a target intervals list (step 1). Then we perform local realignment for the target intervals using IndelRealigner (step 2).
- Introduction
- Create target intervals list using RealignerTargetCreator
- Realign reads using IndelRealigner
- Some additional considerations
- Related resources
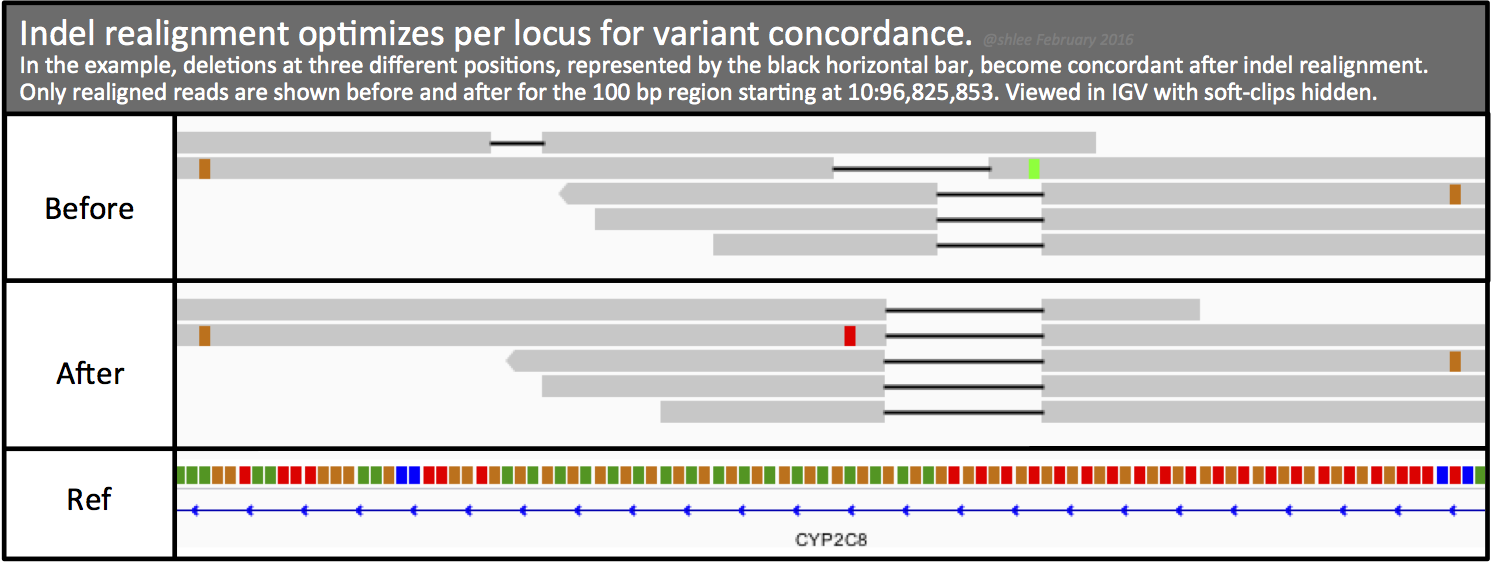
Genome aligners can only consider each read independently, and the scoring strategies they use to align reads relative to the reference limit their ability to align reads well in the presence of indels. Depending on the variant event and its relative location within a read, the aligner may favor alignments with mismatches or soft-clips instead of opening a gap in either the read or the reference sequence. In addition, the aligner's scoring scheme may use arbitrary tie-breaking, leading to different, non-parsimonious representations of the event in different reads.
In contrast, local realignment considers all reads spanning a given position. This makes it possible to achieve a high-scoring consensus that supports the presence of an indel event. It also produces a more parsimonious representation of the data in the region .
This two-step indel realignment process first identifies such regions where alignments may potentially be improved, then realigns the reads in these regions using a consensus model that takes all reads in the alignment context together.
- Installed GATK tools
- Coordinate-sorted and indexed BAM alignment data
- Reference sequence, index and dictionary
- An optional VCF file representing population variants, subset for indels
- To download the reference, open ftp://ftp.broadinstitute.org/bundle/b37/ in your browser. Leave the password field blank. Download the following three files (~860 MB) to the same folder:
human_g1k_v37_decoy.fasta.gz,.fasta.fai.gz, and.dict.gz. This same reference is available to load in IGV. Click tutorial_7156.tar.gz to download the tutorial data. The data is human paired 2x150 whole genome sequence reads originally aligning at ~30x depth of coverage. The sample is a PCR-free preparation of the NA12878 individual run on the HiSeq X platform. I took the reads aligning to a one Mbp genomic interval (10:96,000,000-97,000,000) and sanitized and realigned the reads (BWA-MEM -M) to the entire genome according to the workflow presented in Tutorial#6483 and marked duplicates using MarkDuplicates according to Tutorial#6747. We expect the alignment to reveal a good proportion of indels given its long reads (~150 bp per read), high complexity (PCR-free whole genome data) and deep coverage depth (30x).
Tutorial download also contains a known indels VCF from Phase 3 of the 1000 Genomes Project subset for indel-only records in the interval 10:96,000,000-97,000,000. These represent consensus common and low-frequency indels in the studied populations from multiple approaches. The individual represented by our snippet, NA12878, is part of the 1000 Genomes Project data. Because of the differences in technology and methods used by the Project versus our sample library, our library has potential to reveal additional variants.
For simplicity, we use a single known indels VCF, included in the tutorial data. For recommended resources, see Article#1247.
In the command, RealignerTargetCreator takes a coordinate-sorted and indexed BAM and a VCF of known indels and creates a target intervals file.
java -jar GenomeAnalysisTK.jar \
-T RealignerTargetCreator \
-R human_g1k_v37_decoy.fasta \
-L 10:96000000-97000000 \
-known INDEL_chr10_1Mb_b37_1000G_phase3_v4_20130502.vcf \
-I 7156_snippet.bam \
-o 7156_realignertargetcreator.intervals
In the resulting file, 7156_realignertargetcreator.intervals, intervals represent sites of extant and potential indels. If sites are proximal, the tool represents them as a larger interval spanning the sites.
- We specify the BAM alignment file with
-I. - We specify the known indels VCF file with
-known. The known indels VCF contains indel records only. Three input choices are technically feasible in creating a target intervals list: you may provide RealignerTargetCreator (i) one or more VCFs of known indels each passed in via
-known, (ii) one or more alignment BAMs each passed in via-Ior (iii) both. We recommend the last mode, and we use it in the example command. We use these same input files again in the realignment step.The tool adds indel sites present in the known indels file and indel sites in the alignment CIGAR strings to the targets. Additionally, the tool considers the presence of mismatches and soft-clips, and adds regions that pass a concentration threshold to the target intervals.
If you create an intervals list using only the VCF, RealignerTargetCreator will add sites of indel only records even if SNPs are present in the file. If you create an intervals list using both alignment and known indels, the known indels VCF should contain only indels. See Related resources.
We include
-L 10:96000000-97000000in the command to limit processing time. Otherwise, the tool traverses the entire reference genome and intervals outside these coordinates may be added given our example7156_snippet.bamcontains a small number of alignments outside this region.- The tool samples to a target coverage of 1,000 for regions with greater coverage.
The first ten rows of 7156_realignertargetcreator.intervals are as follows. The file is a text-based one-column list with one interval per row in 1-based coordinates. Header and column label are absent. For an interval derived from a known indel, the start position refers to the corresponding known variant. For example, for the first interval, we can zgrep -w 96000399 INDEL_chr10_1Mb_b37_1000G_phase3_v4_20130502.vcf for details on the 22bp deletion annotated at position 96000399.
10:96000399-96000421 10:96002035-96002036 10:96002573-96002577 10:96003556-96003558 10:96004176-96004177 10:96005264-96005304 10:96006455-96006461 10:96006871-96006872 10:96007627-96007628 10:96008204
To view intervals on IGV, convert the list to 0-based BED format using the following AWK command. The command saves a new text-based file with .bed extension where chromosome, start and end are tab-separated, and the start position is one less than that in the intervals list.
awk -F '[:-]' 'BEGIN { OFS = "\t" } { if( $3 == "") { print $1, $2-1, $2 } else { print $1, $2-1, $3}}' 7156_realignertargetcreator.intervals > 7156_realignertargetcreator.bed
In the following command, IndelRealigner takes a coordinate-sorted and indexed BAM and a target intervals file generated by RealignerTargetCreator. IndelRealigner then performs local realignment on reads coincident with the target intervals using consenses from indels present in the original alignment.
java -Xmx8G -Djava.io.tmpdir=/tmp -jar GenomeAnalysisTK.jar \
-T IndelRealigner \
-R human_g1k_v37_decoy.fasta \
-targetIntervals 7156_realignertargetcreator.intervals \
-known INDEL_chr10_1Mb_b37_1000G_phase3_v4_20130502.vcf \
-I 7156_snippet.bam \
-o 7156_snippet_indelrealigner.bam

- The
-targetIntervalsfile from RealignerTargetCreator, with extension.intervalsor.list, is required. See section 1 for a description. - Specify each BAM alignment file with
-I. IndelRealigner operates on all reads simultaneously in files you provide it jointly. - Specify each optional known indels VCF file with
-known. - For joint processing, e.g. for tumor-normal pairs, generate one output file for each input by specifying
-nWayOutinstead of-o. By default, and in this command, IndelRealigner applies the
USE_READSconsensus model. This is the consensus model we recommend because it balances accuracy and performance. To specify a different model, use the-modelargument. TheKNOWNS_ONLYconsensus model constructs alternative alignments from the reference sequence by incorporating any known indels at the site, theUSE_READSmodel from indels in reads spanning the site and theUSE_SWmodel additionally from Smith-Waterman alignment of reads that do not perfectly match the reference sequence.The
KNOWNS_ONLYmodel can be sufficient for preparing data for base quality score recalibration. It can maximize performance at the expense of some accuracy. This is the case only given the known indels file represents common variants for your data. If you specify-model KNOWNS_ONLYbut forget to provide a VCF, the command runs but the tool does not realign any reads.If you encounter out of memory errors, try these options. First, increase max java heap size from
-Xmx8G. To find a system's default maximum heap size, typejava -XX:+PrintFlagsFinal -version, and look forMaxHeapSize. If this does not help, and you are jointly processing data, then try running indel realignment iteratively on smaller subsets of data before processing them jointly.- IndelRealigner performs local realignment without downsampling. If the number of reads in an interval exceeds the 20,000 default threshold set by the
-maxReadsparameter, then the tool skips the region. - The tool has two read filters, BadCigarFilter and MalformedReadFilter. The tool processes reads flagged as duplicate.
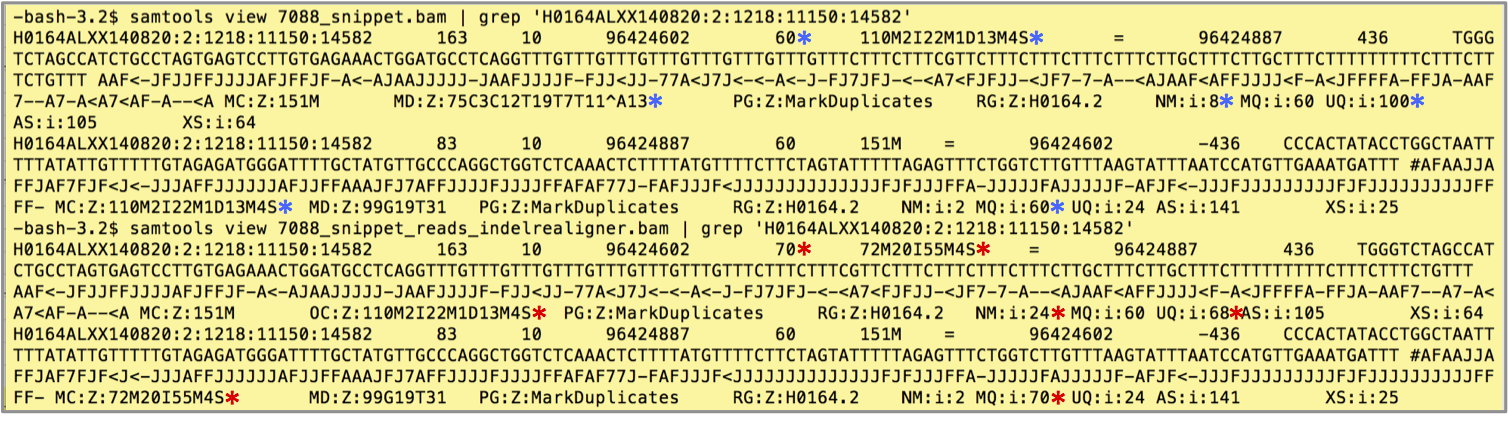
For our example data,194 alignment records realign for ~89 sites. These records now have the OC tag to mark the original CIGAR string. We can use the OC tag to pull out realigned reads and instructions for this are in section 4. The following screenshot shows an example pair of records before and after indel realignment. We note seven changes with asterisks, blue for before and red for after, for both the realigned read and for its mate.
{kind=link}
{kind=link}
Changes to the example realigned record:
- MAPQ increases from 60 to 70. The tool increases each realigned record's MAPQ by ten.
- The CIGAR string, now
72M20I55M4S, reflects the realignment containing a 20bp insertion. - The OC tag retains the original CIGAR string (OC:Z:110M2I22M1D13M4S) and replaces the MD tag that stored the string for mismatching positions.
- The NM tag counts the realigned record's mismatches, and changes from 8 to 24.
Changes to the realigned read's mate record:
- The MC tag updates the mate CIGAR string (to MC:Z:72M20I55M4S).
- The MQ tag updates to the new mapping quality of the mate (to MQ:i:70).
- The UQ tag updates to reflect the new Phred likelihood of the segment, from UQ:i:100 to UQ:i:68.
RealignerTargetCreator documentation has a -maxInterval cutoff to drop intervals from the list if they are too large. This is because increases in number of reads per interval quadratically increase the compute required to realign a region, and larger intervals tend to include more reads. By the same reasoning, increasing read depth, e.g. with additional alignment files, increases required compute.
Our tutorial's INDEL_chr10_1Mb_b37_1000G_phase3_v4_20130502.vcf contains 1168 indel-only records. The following are metrics on intervals created using the three available options.
#intervals avg length basepair coverage VCF only 1161 3.33 3,864 BAM only 487 15.22 7,412 VCF+BAM 1151 23.07 26,558
You can project the genomic coverage of the intervals as a function of the interval density (number of intervals per basepair) derived from varying the known indel density (number of indel records in the VCF). This in turn allows you to anticipate compute for indel realignment. The density of indel sites increases the interval length following a power law (y=ax^b). The constant (a) and the power (b) are different for intervals created with VCF only and with VCF+BAM. For our example data, these average interval lengths are well within the length of a read and minimally vary the reads per interval and thus the memory needed for indel realignment.
- See the Best Practice Workflow and click on the flowchart's
Realign Indelsicon for best practice recommendations and links including to a 14-minute video overview. - See Article#1247 for guidance on using VCF(s) of known variant sites.
- To subset realigned reads only into a valid BAM, as shown in the screenshots, use
samtools view 7088_snippet_indelrealigner.bam | grep 'OC' | cut -f1 | sort > 7088_OC.txtto create a list of readnames. Then, follow direction in blogpost SAM flags down a boat on how to create a valid BAM using FilterSamReads. - See discussion on multithreading for options on speeding up these processes. The document titled How can I use parallelism to make GATK tools run faster? gives two charts: (i) the first table relates the three parallelism options to the major GATK tools and (ii) the second table provides recommended configurations for the tools. Briefly, RealignerTargetCreator runs faster with increasing
-ntthreads, while IndelRealigner shows diminishing returns for increases in scatter-gather threads provided by Queue. See blog How long does it take to run the GATK Best Practices? for a breakdown of the impact of threading and CPU utilization for Best Practice Workflow tools. - See DePristo et al's 2011 Nature Genetics technical report for benchmarked effects of indel realignment as well as for the mathematics behind the algorithms.
- See Tutorial#6517 for instructions on creating a snippet of reads corresponding to a genomic interval. For your research aims, you may find testing a small interval of your alignment and your choice VCF, while adjusting parameters, before committing to processing your full dataset, is time well-invested.
- The tutorial's PCR-free 2x150 bp reads give enough depth of coverage (34.67 mean and 99.6% above 15) and library complexity to allow us the confidence to use aligner-generated indels in realignment. Check alignment coverage with DepthofCoverage for WGS or DiagnoseTargets for WES.
- See SelectVariants to subset out indel calls using the
-selectType INDELoption. Note this excludes indels that are part of mixed variant sites (see FAQ). Current solutions to including indels from mixed sites involves the use of JEXL expressions, as discussed here. Current solutions to selecting variants based on population allelic frequency (AF), as we may desire to limit our known indels to those that are more common than rare for more efficient processing, are discussed in two forum posts (1,2). - See Tutorial#6491 for basic instructions on using the Integrative Genomics Viewer (IGV).